
Ensuring steroid-free food supplements
Identification of steroids in pharmaceuticals and food supplements with LCMS-8045
Anabolic steroids are often encountered in samples of nutritional supplements and pharmaceutical preparations, either due to doping-related activities [1] or cross-contamination issues during production [2]. Developing methods to identify steroids in such samples can be challenging, since the method should be able to monitor numerous steroids with large differences in terms of polarity. Compounds to be monitored comprise relatively polar steroids such as anastrozole as well as low polarity steroid esters which have been occurring with increasing frequency in food supplement samples [3].
The method developed includes testing for testosterone caproate and testosterone isocaproate, a challenging set of isomers which were partially separated (Resolution 0.6 as per Eur.Ph.) enabling the analytical method to identify which of the two isomers is contained in the sample. With respect to tibolone, three MRM transitions are reported with no need to resort to derivatization, which is usually the case with tibolone and its metabolites. Finally, a comprehensive rationale is provided for choosing MRM transitions for steroid esters, based on fragments of the free steroid.
Instrumentation, method parameters and sample preparation
The experiments were conducted with Nexera X2 UHPLC system coupled to LCMS-8045 triple quadrupole mass spectrometer. The system consisted of two LC-30AD pumps, a SIL-30AC autosampler and a CTO-20AC column oven with a flow selection valve. The valve was used to divert mobile phase flow to waste during the first 1.5 minutes and avoid contamination of the mass spectrometer with early-eluting matrix components.
Chromatographic separation was achieved with Zorbax SB-C18 Rapid Resolution HT, 50 x 4.6 mm, 3.5 µm (part number 835975-902) maintained at 40 °C. The mobile phase was a mixture of 0.1 % aqueous formic acid and methanol, with gradient elution and a flowrate of 0.4 mL/min. This set of parameters resulted in retention times ranging from 2.4 – 24.0 minutes and a total runtime of 31 minutes including the equilibration step.
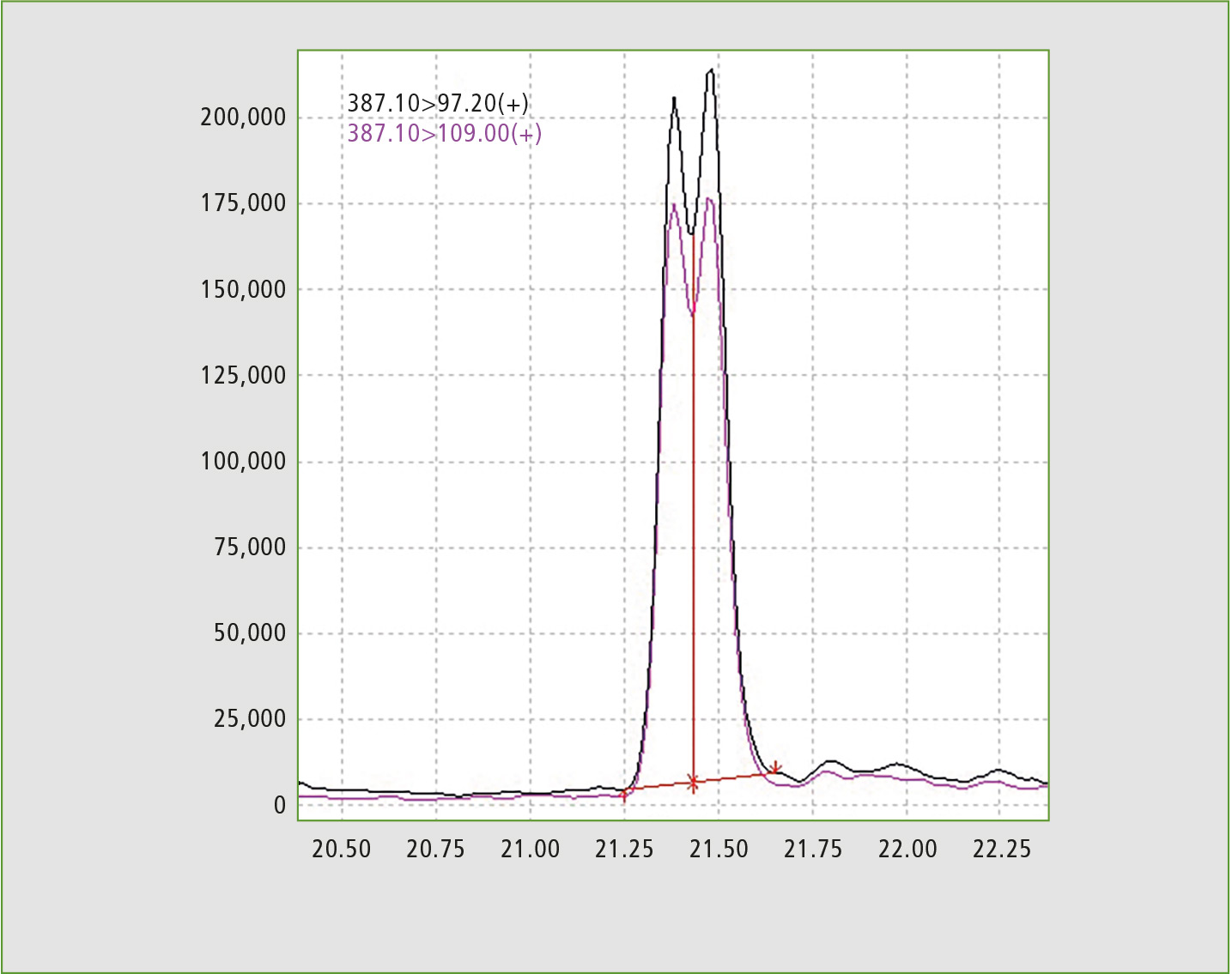 Figure 1: Chromatogram of standard solution, exhibiting partial separation of testosterone caproate and testosterone isocaproate
Figure 1: Chromatogram of standard solution, exhibiting partial separation of testosterone caproate and testosterone isocaproate
Liquid chromatography
Method development strategy aimed at including a broad spectrum of compounds with respect to polarity while ensuring a reasonable runtime, with a total of 16 steroid esters being included in the analytical method. Regarding the mobile phase flowrate, 0.5 mL/min and 0.4 mL/min were tested, with the latter being chosen due to increased sensitivity.
Two mobile phase compositions were tested with gradient elution, the first being a mixture of 1 mM aqueous ammonium acetate/1 mM methanolic ammonium acetate and the second being a mixture of 0.1 % aqueous formic acid/ methanol. The second mobile phase was chosen to ensure simpler preparation procedure. The change in mobile phase acidity using formic acid appeared to have minor effects on retention time of the compounds since early eluting compounds exhibited approximately 20 % higher retention, whereas late-eluting compounds exhibited approximately 10 % higher retention.
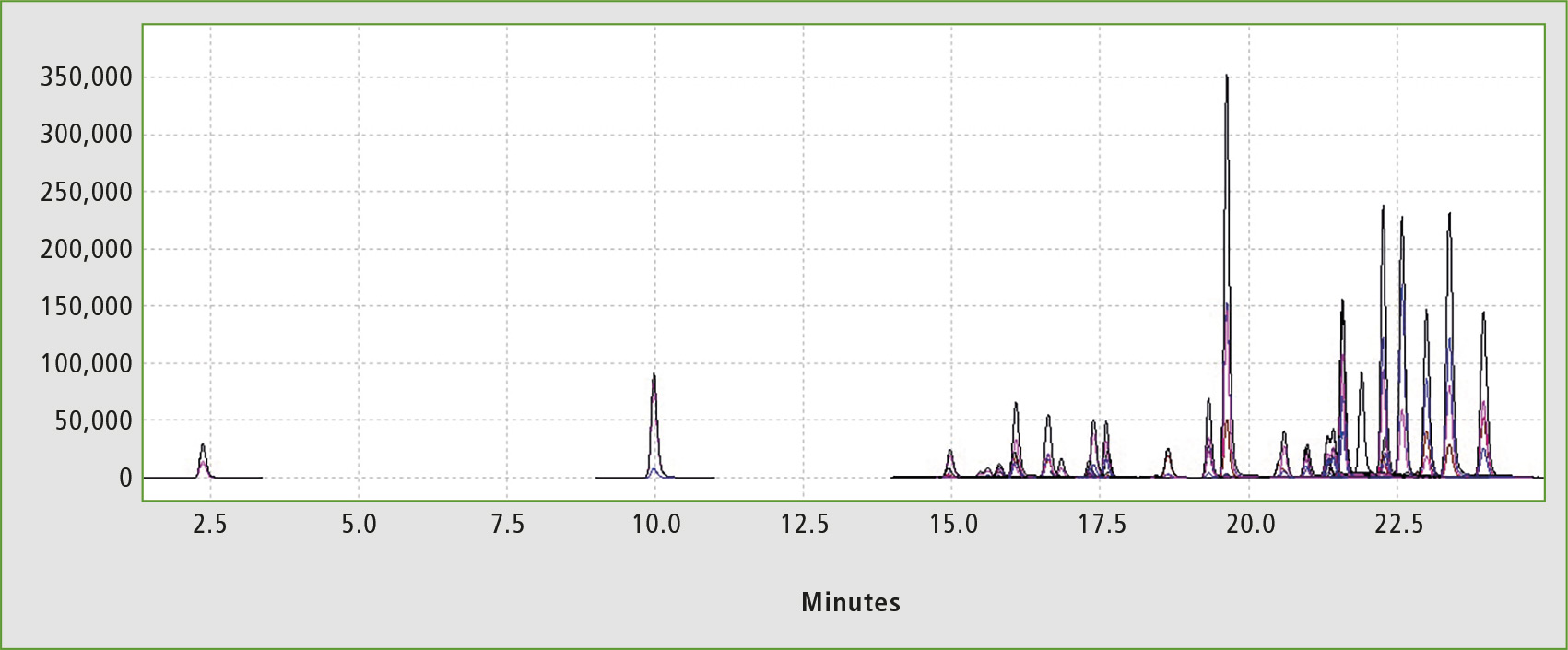 Figure 2: Chromatogram of the standard solution
Figure 2: Chromatogram of the standard solution
Two analytical columns were tested, namely one Zorbax SB-C18 Rapid Resolution HT, 50 x 2.1 mm, 1.8 µm (part number 822700-902) and one Zorbax SB-C18 Rapid Resolution HT, 50 x 4.6 mm, 3.5 µm (part number 835975-902). Given that the first column contained sub-2 µm particles, considerably shorter sample runtime was expected, however comparable retention times were obtained with both columns. Since the UHPLC column was prone to overpressure and clogging after a few analyses, the 3.5 µm column was preferred for the application, with an injection volume of 2 µL.
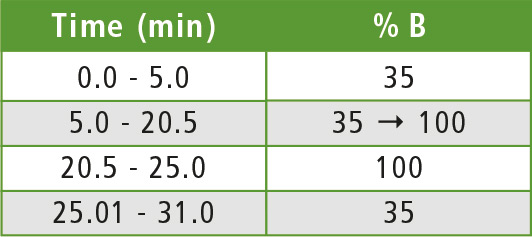 Table 1: Gradient elution program
Table 1: Gradient elution program
Autosampler temperature was initially set to 4 °C to ensure stability of the samples. Because heavy precipitation of matrix components was observed when the vials were exposed to 4 °C, the autosampler was kept at room temperature and 24-hour stability was established for the analytes.
Sample preparation
In order to keep sample pretreatment simple, effortless steps were employed. The solid dosage form samples were homogenized and 100 mg was weighed in a 10 mL volumetric flask. Due to the high solubility of the compounds in methanol, the homogenized sample was dissolved in methanol to ensure high recovery for all compounds (Test Solution 1). Four Test Solutions (TS) were then prepared (TS 2-5) for injection into the instrument.
Because matrix components contained in TS1 were not always soluble in mobile phase, 1.0 mL of this solution was filtered through a 0.22 µm nylon filter and diluted to volume with mobile phase to avoid precipitation inside the column (Test Solution 2, dilution 1:10). 1.0 mL each of TS2, TS3, and TS4 was transferred to 10 mL volumetric flasks and diluted to volume with mobile phase (TS3, dilution 1:102; TS4, dilution 1:103; TS5, dilution 1:104). 2 µL of TS3 and TS4 were injected into the instrument. TS2 would be injected if low intensity peaks were obtained with TS3 and TS4. TS5 would be injected if high intensity peaks were obtained with TS3 and TS4.
Mass spectrometry
The compounds to be determined were divided into six groups of similar structure: testosterone and testosterone esters, trenbolone esters, nandrolone and nandrolone esters, methenolone esters, boldenone esters and drostanolone esters. The fragments of steroid esters of the same group were expected to be, to a certain extent, common, which was confirmed during the initial mass spectrometry experiments as can be seen in table 2. Therefore, common fragments mentioned in table 2 may be used to choose MRM transitions for other steroid esters, based on fragments of the free steroid.
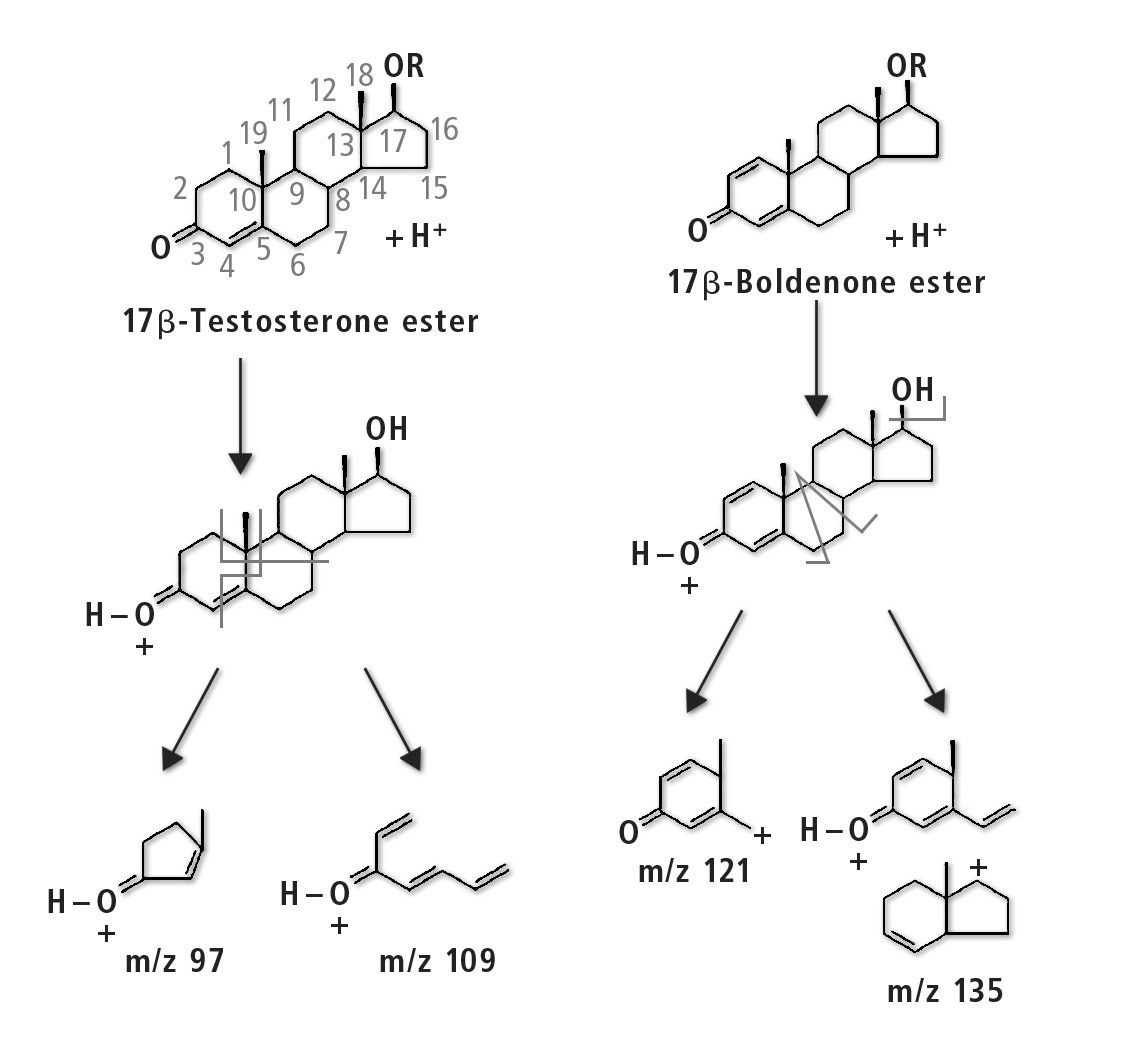 Figure 3: Fragmentation pathway of testosterone group and boldenone group of compounds [3]
Figure 3: Fragmentation pathway of testosterone group and boldenone group of compounds [3]
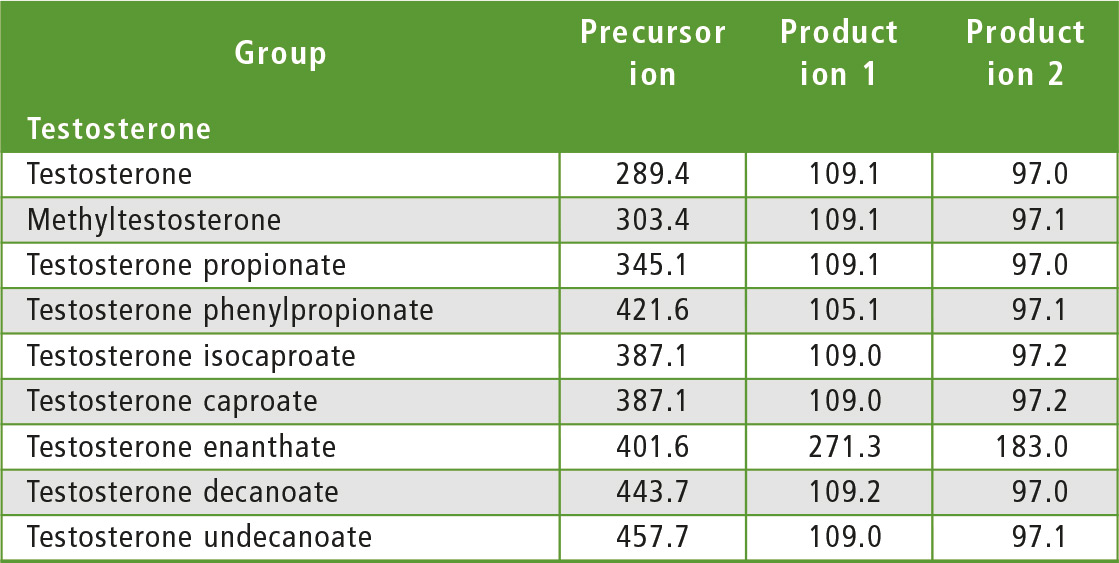
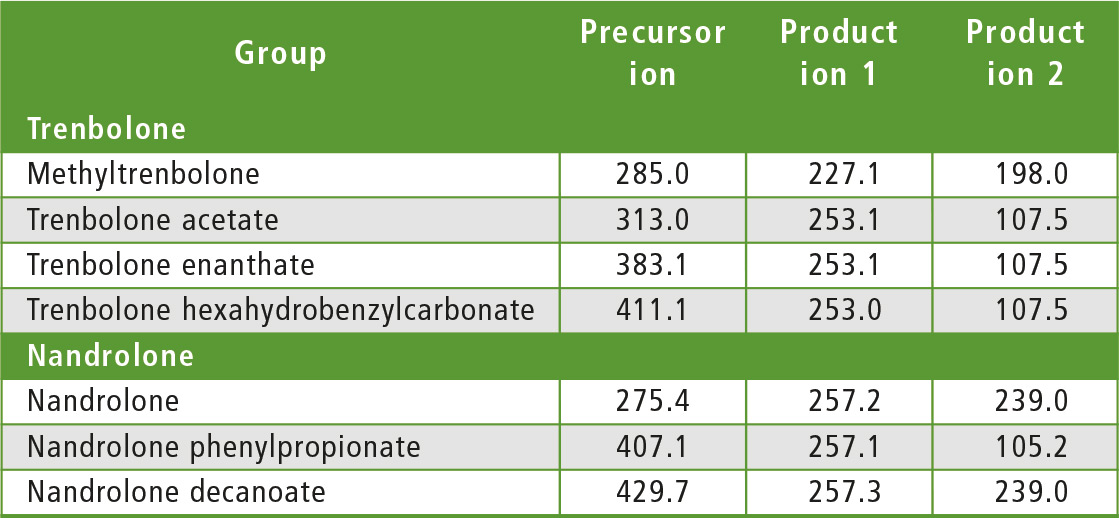
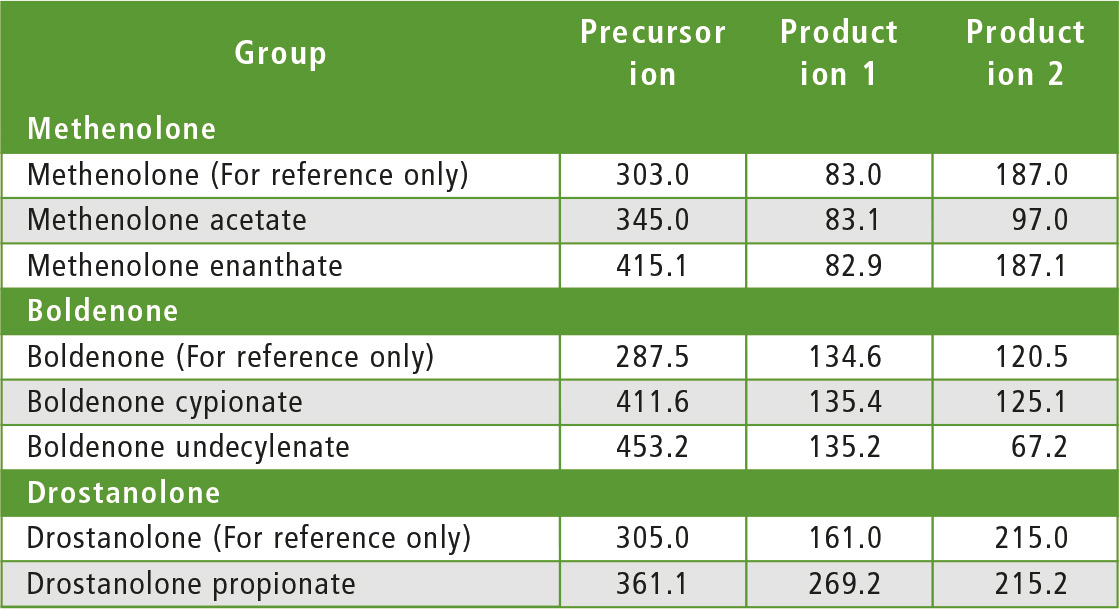 Table 2: Rationale for the choice of MRM transitions
Table 2: Rationale for the choice of MRM transitions
With respect to testosterone group, m/z 109 was used for quantitation and m/z 97 was used for confirmation. Both ions are characteristic fragments of testosterone and their use is proven in the literature [1-3]. Moreover, because the two ions correspond to fragments of the steroid core of the molecule, their applicability to all testosterone esters was investigated and confirmed. A similar rationale was followed for the group of nandrolone and nandrolone esters, m/z 257 and m/z 239. For the group of trenbolone esters, m/z 253 and m/z 107 were used. For the group of boldenone esters, m/z 135 was applied, and for drostanolone propionate, m/z 215 was used.
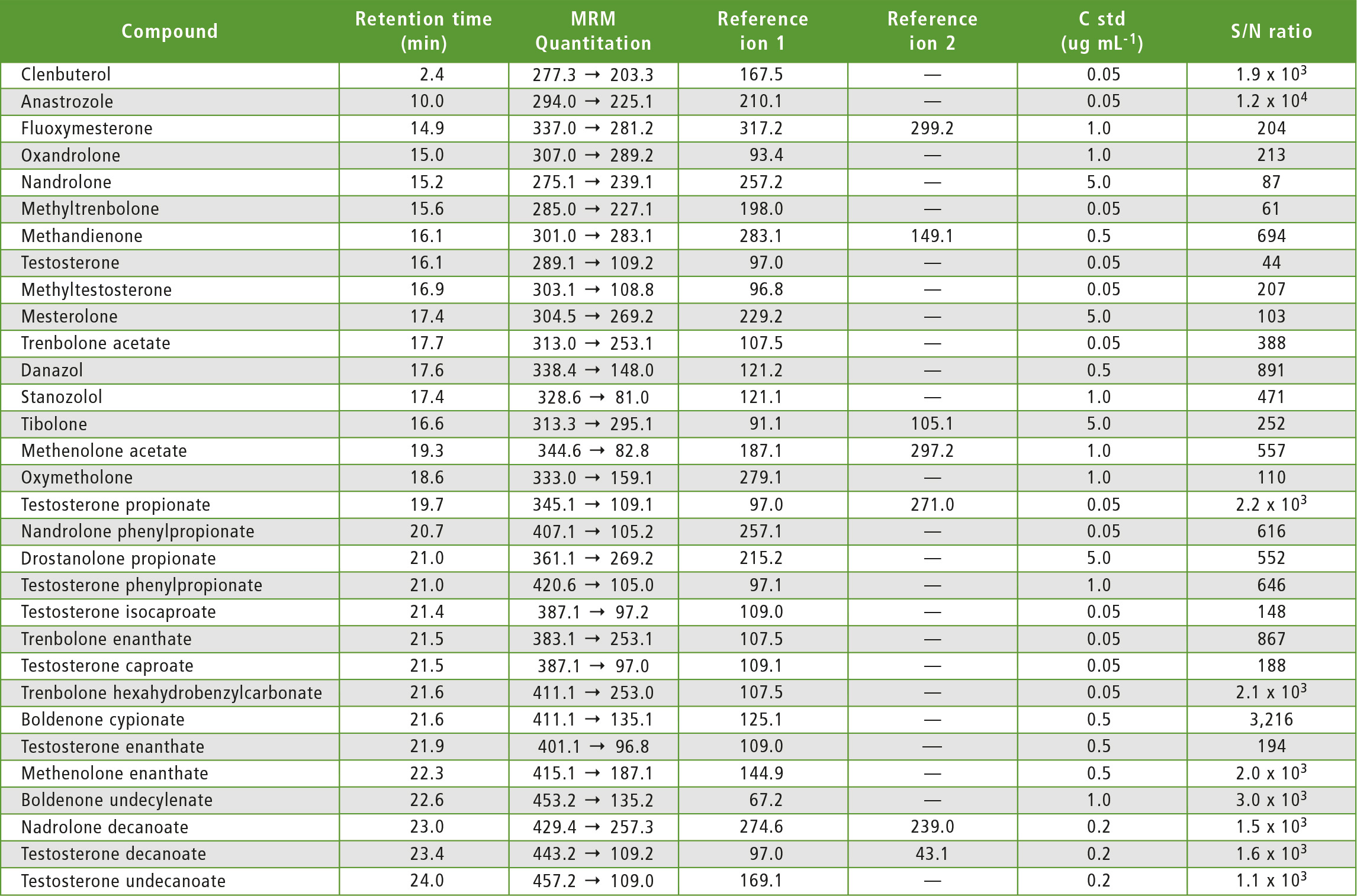 Table 3: MRM transitions for each compound
Table 3: MRM transitions for each compound
The optimized mass spectrometry parameters were:
Positive ionization: (ESI capillary voltage set to -4 kV)
Heat block temperature: 400 °C
Desolvation line temperature: 250 °C
Heating gas temperature: 350 °C
Drying gas: 10.0 L/ min
Nebulizing gas: 3.0 L/min
Heating gas: 10.0 L/min
Dwell time was set to 20 ms, except for:
Nandrolone: (100 ms)
Fluoxymesterone: (50 ms)
Methyltrenbolone: (150 ms)
Methenolone acetate: (50 ms)
Drostanolone propionate: (150 ms).
Conclusions
An analytical method was developed to identify 31 steroids in pharmaceutical preparations and food supplements. This is the first method to identify testosterone caproate in the presence of its isomer, testosterone isocaproate, and vice versa. Key parameters that affect the analysis of all 31 steroids have been identified and taken into account.

Authors
Dr. Gerasimos Liapatas, Dr. Manos Barbounis
Applications Department of N.Asteriadis S.A. 31 Dervenion Str. & Poseidonos Str., 144
51 Metamorfossi – Athens, Greece
mb@asteriadis.gr
Literature
[1] C. Borges, N. Miller, M. Shelby, M. Hansen, C. White, M.H. Slawson, K. Monti, D.J. Crouch, Analysis of a challenging subset of World Anti Doping Agency-Banned steroids and anti-estrogens by LC-MS-MS, Journal of Analytical Toxicology, 31 (2007) 125-131.
[2] S. Fekete, J. Fekete, K. Ganzler, Validated UPLC method for the fast and sensitive determination of steroid residues in support of cleaning validation in formulation area, Journal of Pharmaceutical and Biomedical analysis, 49 (2009) 833-838.
[3] M. Thevis, Y. Schrader, A. Thomas, G. Sigmund, H. Geyer, W. Schanzer, Analysis of confiscated black market drugs using chromatographic and mass spectrometric approaches, Journal of Analytical Toxicology 2008;32:232-240.